Login
Welcome back! Please enter your details.
or
Don't have an account? Register here
Create Account
Join MedMentorEdu and start your medical journey.
or
Already have an account? Login here
Enhance your knowledge with our comprehensive guide and curated study materials.

Hamartoneoplastic syndromes are genetic disorders characterized by the presence of hamartomas, which are benign growths composed of disorganized but mature tissues normally present at the site.
These syndromes are associated with an increased risk of malignancies.
Multiple organ systems may be involved, including:
• Skin
• Nervous system
• Bones
• Caused by mutations in tumor suppressor genes, leading to loss of normal regulation of cell growth.
• These mutations allow uncontrolled cellular proliferation, resulting in hamartomas and increased tumor risk.
• Many hamartoneoplastic syndromes belong to the group of neurocutaneous disorders, including neurofibromatosis.
Neurofibromatoses are a group of autosomal dominant genetic disorders characterized by:
• Nerve sheath tumors (neurofibromas)
• Café-au-lait macules
• Neurological abnormalities
Two major types are recognized:
• Neurofibromatosis Type 1 (NF1) — von Recklinghausen disease
• Neurofibromatosis Type 2 (NF2) — central neurofibromatosis
A common autosomal dominant disorder affecting the skin, nervous system, and skeletal system.
• Prevalence: approximately 1 in 3,000 births
• Caused by mutations in the NF1 gene located on chromosome 17q11.2.
• The gene encodes neurofibromin, a tumor suppressor protein.
• Loss of neurofibromin results in uncontrolled proliferation of Schwann cells and other neural crest–derived cells.
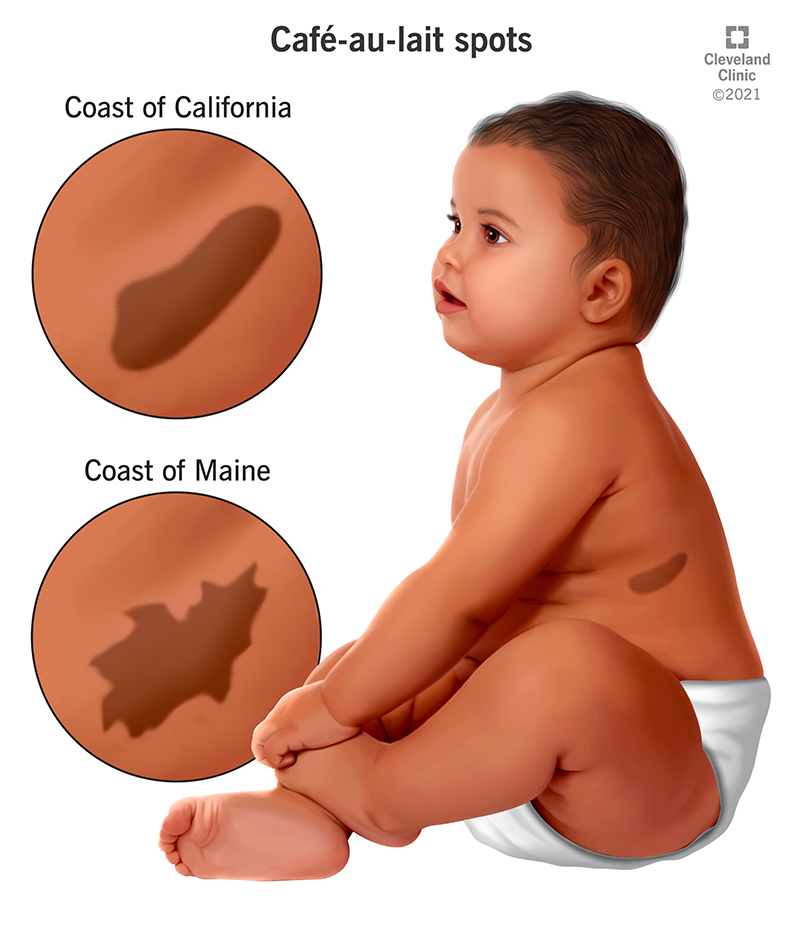
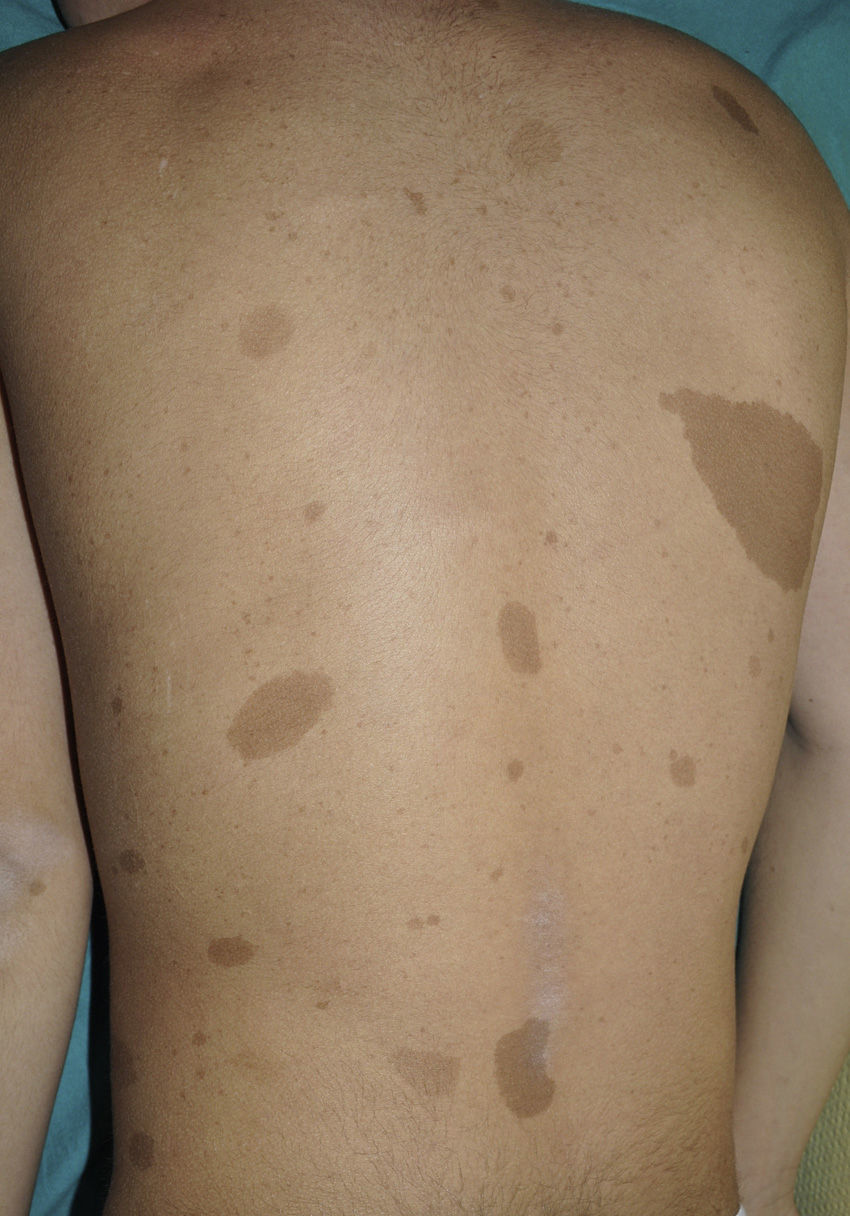
• Café-au-lait macules
• ≥6 lesions required for diagnostic criteria
• Size criteria:
• >5 mm before puberty
• >15 mm after puberty
• Axillary and inguinal freckling
• Known as Crowe’s sign
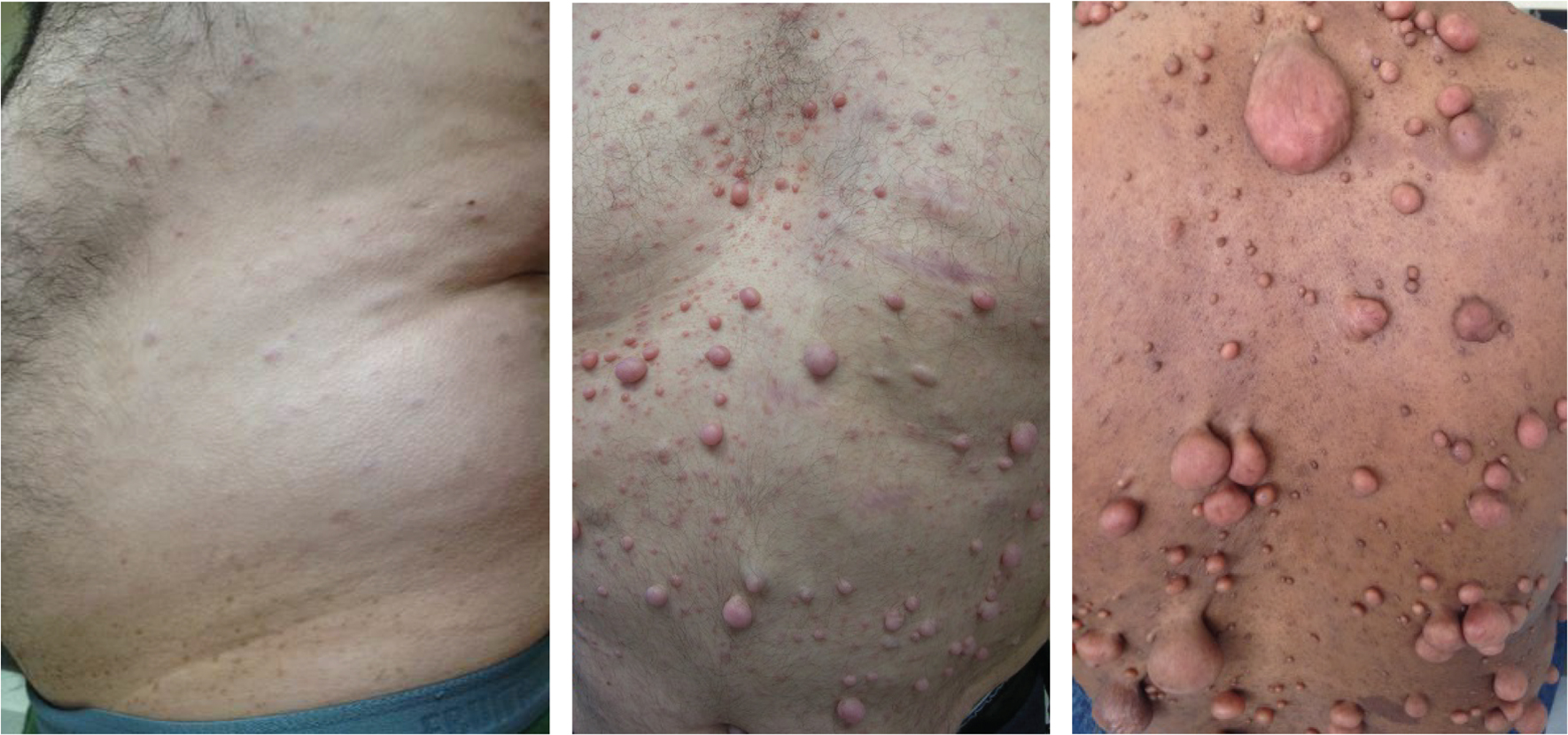
• Multiple neurofibromas
• Cutaneous
• Subcutaneous
• Plexiform
• Glomus tumors
• Painful subungual tumors
• Optic pathway gliomas
• Occur in 15–20% of patients
• Learning disabilities and ADHD
• Common in affected children
• Seizures
• Macrocephaly
• Sphenoid dysplasia
• Can lead to pulsating exophthalmos
• Pseudoarthrosis
• Most commonly affects the tibia
• Scoliosis
• Lisch nodules
• Iris hamartomas
• Considered pathognomonic for NF1
• Glaucoma
• Visual impairment due to optic gliomas
• Hypertension
• May result from renal artery stenosis
• Or pheochromocytoma
• Malignancy risk
• Malignant peripheral nerve sheath tumors (MPNST)
• Gliomas
• Rhabdomyosarcoma
Diagnosis requires two or more of the following:
• ≥6 café-au-lait macules (size criteria depend on age)
• Axillary or inguinal freckling
• ≥2 neurofibromas or 1 plexiform neurofibroma
• Optic glioma
• ≥2 Lisch nodules
• Characteristic skeletal abnormalities
(sphenoid dysplasia or pseudoarthrosis)
• First-degree relative with NF1
There is no definitive cure, and management focuses on monitoring and treating complications.
• Regular ophthalmologic screening for optic gliomas
• Monitoring skeletal abnormalities
• Surgical removal of symptomatic or disfiguring neurofibromas
• Antihypertensive therapy for renovascular hypertension
• Psychological and educational support for learning disabilities
The prognosis of NF1 is highly variable.
• Some individuals experience mild cutaneous disease, while others develop significant neurological and skeletal complications.
• Increased malignancy risk, particularly malignant peripheral nerve sheath tumors, contributes to reduced life expectancy in severe cases.


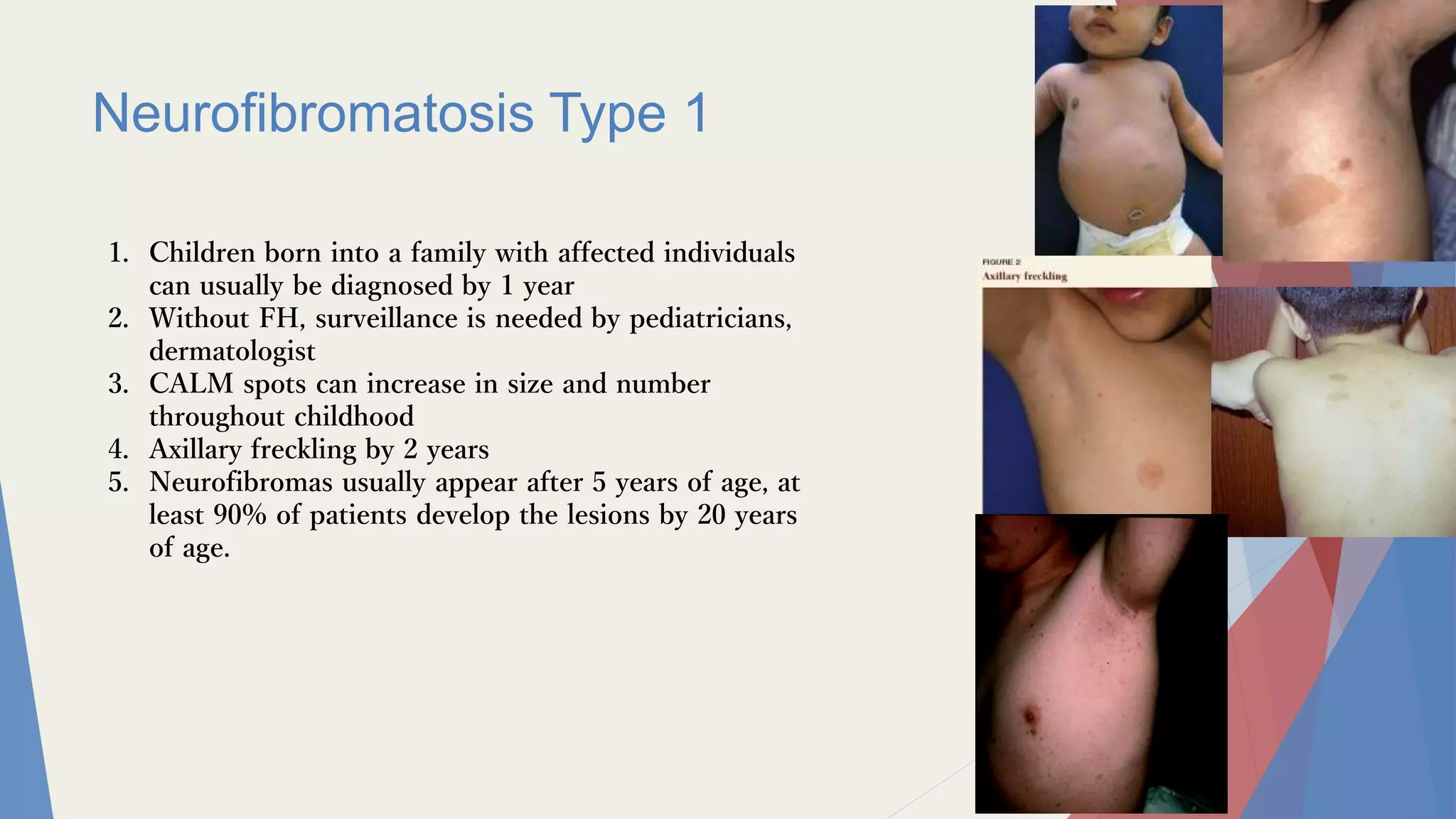
Neurofibromatosis Type 1 (NF1) is associated with an increased risk of hematological malignancies, particularly juvenile chronic myeloid leukemia (JCML).
Juvenile xanthogranuloma (JXG), a benign histiocytic skin lesion, occurs more frequently in children with NF1 and may indicate a higher risk of developing JCML.
• Mutation of the NF1 gene causes loss of neurofibromin, a tumor suppressor protein.
• This results in dysregulation of the RAS signaling pathway, promoting uncontrolled cellular proliferation.
• The abnormal signaling increases susceptibility to myeloproliferative disorders, including JCML.
• JXG lesions consist of:
• Histiocytes
• Foamy macrophages
• These lesions are commonly observed in infants and young children.
• A benign non-Langerhans cell histiocytosis occurring mainly in early childhood.
Skin Lesions
• Yellowish-orange papules or nodules
• Common sites:
• Head
• Neck
• Upper trunk
• Lesions are usually solitary but may be multiple.
• Most cases resolve spontaneously over time.
• Presence of JXG in patients with NF1 increases the risk of JCML.
• An aggressive myeloproliferative disorder of early childhood.
• Pallor
• Hepatosplenomegaly
• Lymphadenopathy
• Easy bruising
• Recurrent infections
Peripheral Blood
• Monocytosis
• Anemia
• Thrombocytopenia
Bone Marrow
• Myeloid hyperplasia
• Absence of the Philadelphia chromosome (Ph−)
• Clinical examination
• Histopathology showing:
• Foamy macrophages
• Touton giant cells
• Peripheral blood smear
• Bone marrow biopsy
• Genetic testing for NF1 mutation if NF1 has not already been diagnosed
• Observation (most lesions resolve spontaneously)
• Surgical excision if lesions are:
• Disfiguring
• Symptomatic
• Hematopoietic Stem Cell Transplantation (HSCT)
• Currently the only curative therapy
• Supportive care
• Blood transfusions
• Antibiotic therapy
• Juvenile Xanthogranuloma
• Excellent prognosis
• Typically self-limiting
• Juvenile Chronic Myeloid Leukemia
• Poor prognosis without HSCT
• Children with NF1 and JXG require careful hematological monitoring due to the increased risk of leukemia.
• Caused by NF1 gene mutation on chromosome 17 → loss of tumor suppressor activity.
• Key clinical triad:
• Café-au-lait macules
• Neurofibromas
• Axillary freckling
• Other complications include:
• Optic gliomas
• Lisch nodules
• Scoliosis
• Hypertension
• Diagnosis based on NIH criteria (≥2 features required).
• Increased risk of malignant peripheral nerve sheath tumors.
• Juvenile xanthogranuloma is more common in children with NF1.
• Presence of JXG may indicate increased risk of JCML.
• JCML is an aggressive leukemia requiring HSCT for cure.
• Although JXG usually resolves spontaneously, NF1 patients with JXG should undergo regular hematologic monitoring.



Segmental neurofibromatosis is a localized form of Neurofibromatosis Type 1 (NF1) in which characteristic lesions are restricted to a specific body segment.
Typical findings include:
• Café-au-lait macules
• Neurofibromas
• Pigmentary abnormalities
Lesions usually follow Blaschko’s lines, reflecting embryonic skin cell migration patterns.
Unlike classic NF1, segmental NF does not follow autosomal dominant inheritance.
• Caused by post-zygotic somatic mutation in the NF1 gene located on chromosome 17q11.2.
• This results in somatic mosaicism, meaning only a subset of body cells carry the mutation.
• Because the mutation occurs after fertilization, the abnormal cells are distributed in patchy or segmental patterns.
• Unilateral distribution of lesions
• Typically does not cross the midline
• Lesions limited to a single dermatome or body segment
Pigmentary Type
• Only café-au-lait macules and freckling
Tumorous Type
• Localized neurofibromas
Mixed Type
• Combination of pigmentary changes and neurofibromas
Hereditary Type
• Mutation involves gonadal mosaicism, allowing transmission of NF1 to offspring
• Clinical examination showing segmental distribution
• Absence of generalized NF1 features
• Genetic testing for NF1 mosaicism may be performed in selected cases.
• Observation in mild localized disease
• Surgical removal of symptomatic neurofibromas
• Regular monitoring if plexiform neurofibromas are present due to risk of malignant transformation.
• Generally good prognosis
• Risk increases if plexiform neurofibromas or malignant peripheral nerve sheath tumors develop.

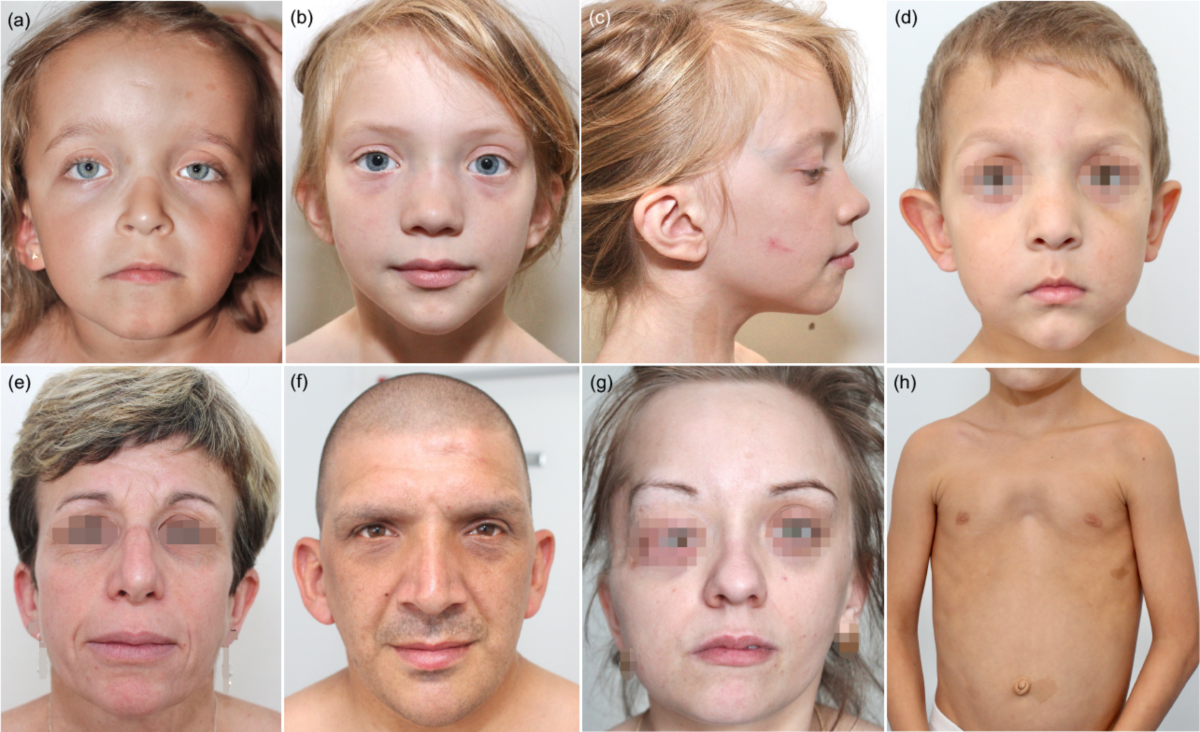
This condition refers to café-au-lait macules occurring in association with congenital pulmonary stenosis.
It may occur as part of Neurofibromatosis–Noonan syndrome or other RASopathies.
RASopathies are a group of disorders caused by mutations affecting the RAS–MAPK signaling pathway.
• Caused by germline mutations affecting the RAS–MAPK pathway.
Common genes involved:
• NF1 gene
• PTPN11 gene
These mutations disrupt cell growth regulation and developmental signaling pathways.
• Café-au-lait macules
• Usually fewer in number than in NF1
• Congenital pulmonary stenosis
• May be valvular or supravalvular
• Noonan-like facial features
• Hypertelorism
• Low-set ears
• Down-slanting palpebral fissures
• Short stature
• Echocardiography
• Used to evaluate severity of pulmonary stenosis
• Genetic testing
• Detection of NF1 or PTPN11 mutations
• Cardiology evaluation is essential.
• Monitoring of pulmonary valve obstruction
• Surgical correction or balloon valvuloplasty may be required in severe cases.
Prognosis largely depends on the severity of pulmonary stenosis and the extent of systemic involvement.
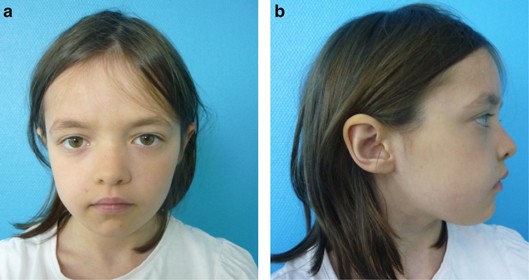
Neurofibromatosis–Noonan syndrome (NFNS) is a genetic disorder showing overlapping features of Neurofibromatosis Type 1 (NF1) and Noonan syndrome (NS).
It belongs to the group of RASopathies, disorders caused by mutations affecting the RAS–MAPK signaling pathway.
• NF1 mutation (most cases)
• Located on chromosome 17q11.2
• PTPN11 mutation (rare cases)
• Gene involved in RAS–MAPK intracellular signaling
These mutations disrupt normal cell growth and developmental pathways.
• Café-au-lait macules
• Axillary and inguinal freckling
• Pulmonary valve stenosis
• One of the most common features inherited from Noonan syndrome
• Short stature
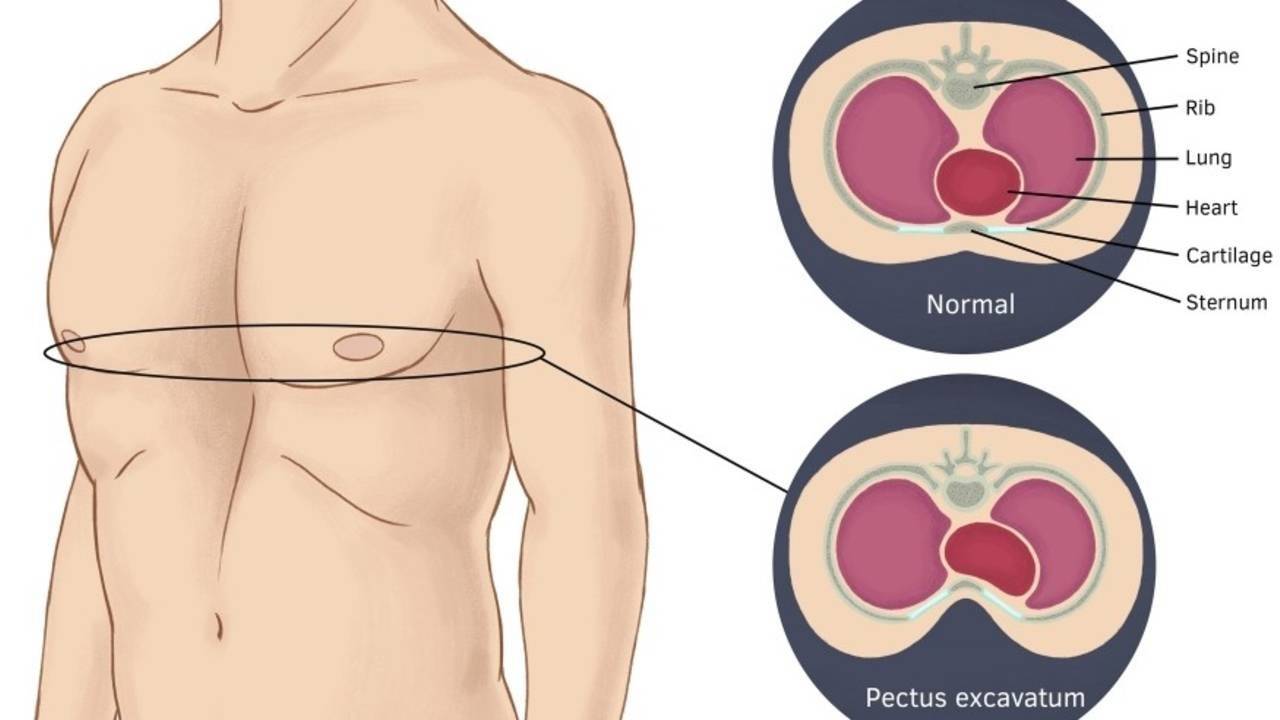
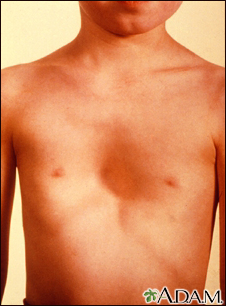
• Pectus deformity
• Pectus excavatum
• Pectus carinatum
• Hypertelorism
• Low-set ears
• Ptosis
• Webbed neck
• Mild intellectual disability may occur
• Usually less severe than in classic NF1
• Genetic testing
• NF1 gene mutation
• PTPN11 mutation
• Echocardiography
• Detects pulmonary stenosis
• Management similar to NF1, with additional cardiology monitoring.
• Regular cardiac evaluation for pulmonary valve stenosis.
• Growth hormone therapy may be considered in severe growth delay.
• Generally better prognosis than classic NF1.
• Requires lifelong monitoring due to potential cardiac and neurological complications.


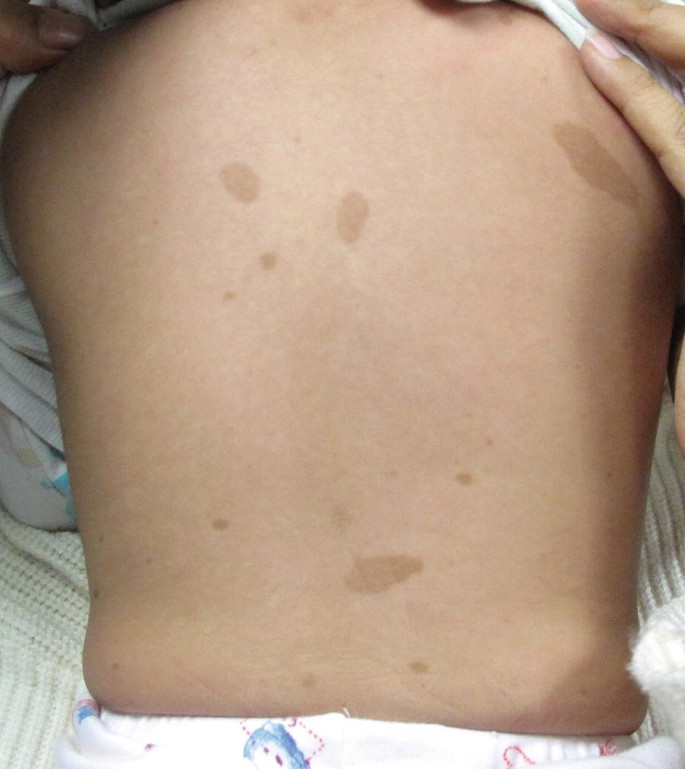

Legius syndrome is a RASopathy with pigmentary features similar to NF1, but without tumor formation.
It presents with café-au-lait macules and freckling, mimicking NF1 but lacking neurofibromas and optic gliomas.
• Caused by mutation in the SPRED1 gene located on chromosome 15q14.
• SPRED1 regulates the RAS–MAPK signaling pathway.
• Mutation leads to abnormal melanocyte proliferation, producing pigmentary lesions.
• Multiple café-au-lait macules
• Axillary and inguinal freckling
• Mild learning disabilities
• Attention or developmental difficulties may occur.
• No neurofibromas
• No optic gliomas
• No Lisch nodules
• No plexiform neurofibromas
• Genetic testing for SPRED1 mutation
• Important to differentiate from NF1, particularly in children with only pigmentary findings.
• Reassurance and observation
• Educational and learning support if developmental difficulties occur.
• Excellent prognosis
• No increased risk of malignancy, unlike NF1.
The strange elegance here is that many of these disorders—NF1, NFNS, Legius syndrome—are variations on the same molecular theme: a slightly misbehaving RAS–MAPK signaling pathway. Think of it as a cellular accelerator pedal stuck halfway down. Different genes tweak different parts of the circuit, producing syndromes that look similar on the skin but behave very differently internally. Dermatology ends up acting like a visible map of molecular signaling errors hidden deep inside the cell.

• Localized NF1 manifestations due to somatic NF1 mosaicism.
• Characterized by unilateral café-au-lait macules and localized neurofibromas.
• Generally good prognosis, unless plexiform neurofibromas or malignant transformation occur.
• Represents an overlap of NF1 and Noonan syndrome features.
• Clinical findings include:
• Café-au-lait macules
• Pulmonary valve stenosis
• Short stature
• Characteristic dysmorphic facial features
• NF1-like disorder with pigmentary findings but without tumor formation.
• Features include:
• Café-au-lait macules
• Axillary or inguinal freckling
Absent features:
• Neurofibromas
• Lisch nodules
• Optic gliomas
• Caused by SPRED1 gene mutation.
• Group of developmental disorders caused by dysregulation of the RAS–MAPK signaling pathway.
Common clinical features include:
• Café-au-lait macules
• Congenital cardiac defects
• Growth retardation
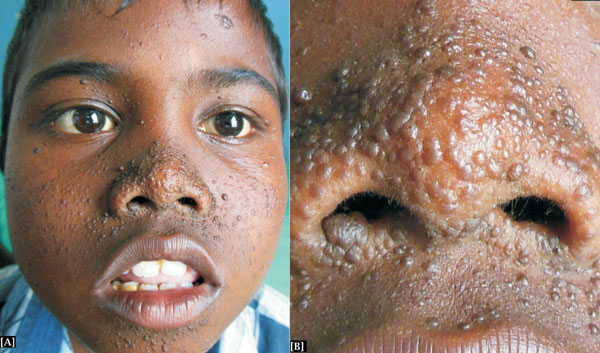

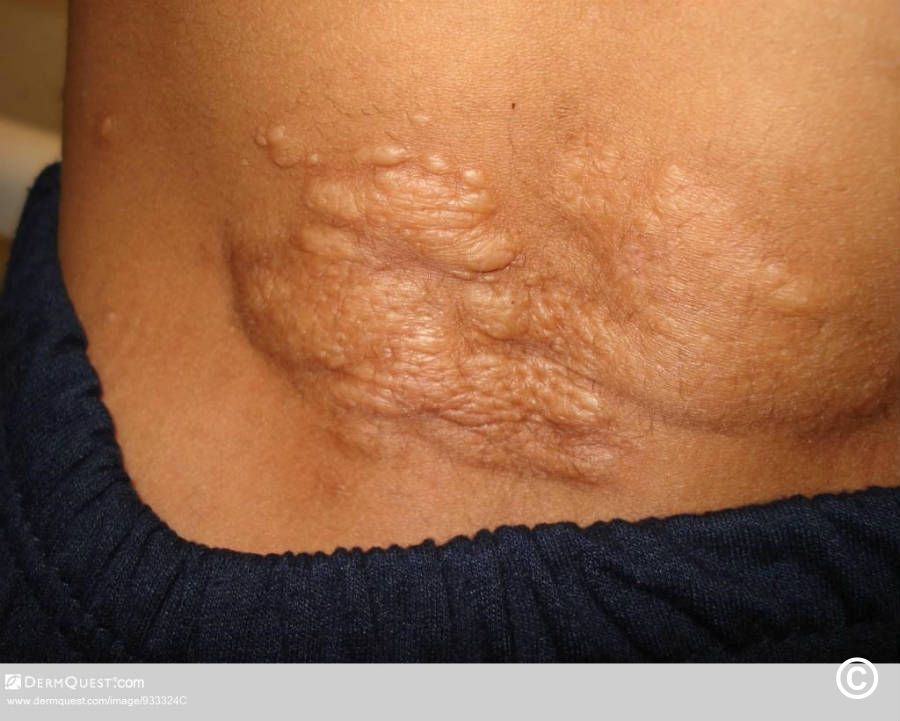

Tuberous sclerosis complex is an autosomal dominant neurocutaneous disorder characterized by the formation of hamartomas in multiple organs, including:
• Skin
• Brain
• Kidneys
• Heart
• Lungs
• Caused by mutations in:
• TSC1 gene (hamartin)
• TSC2 gene (tuberin)
• These genes regulate the mTOR signaling pathway, which controls cell growth and proliferation.
• Mutations lead to uncontrolled cellular proliferation and hamartoma formation.
• Facial angiofibromas (adenoma sebaceum)
• Pink papules on nose and cheeks
• Shagreen patches
• Thickened leathery plaques, typically on the lumbosacral region
• Hypomelanotic macules (Ash-leaf spots)
• Best visualized under Wood’s lamp
• Ungual or periungual fibromas
• Flesh-colored papules around nails
• Forehead plaques
• Rough-textured plaques on the forehead
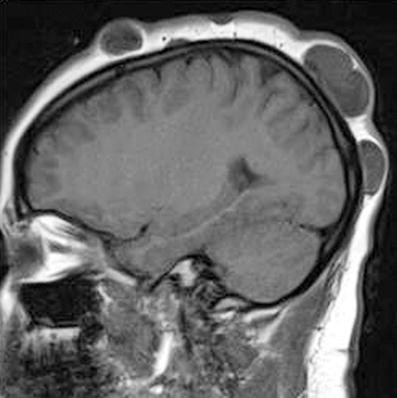
• Cortical tubers
• Associated with developmental delay and seizures
• Subependymal nodules (SENs)
• May calcify over time
• Subependymal giant cell astrocytomas (SEGAs)
• Can cause hydrocephalus
• Renal angiomyolipomas (AMLs)
• Risk of life-threatening hemorrhage
• Renal cysts
• Polycystic kidney disease
• Associated with TSC2–PKD1 deletion
• Cardiac rhabdomyomas
• Most common cardiac tumor in infants
• Often regress spontaneously after birth
• Lymphangioleiomyomatosis (LAM)
• Progressive cystic lung disease
• More common in adult females
Definite TSC
• 2 major criteria, or
• 1 major + 2 minor criteria
Major criteria include:
• Facial angiofibromas
• Shagreen patches
• Cortical tubers
• Renal angiomyolipomas
• Subependymal giant cell astrocytomas
Minor criteria include:
• Dental pits
• Confetti skin lesions
• Multiple renal cysts
• Everolimus (mTOR inhibitor)
• Used for SEGAs and renal angiomyolipomas
• Antiepileptic drugs
• Vigabatrin preferred for infantile spasms
• Regular imaging surveillance
• Brain MRI
• Renal imaging
• Cardiac evaluation
• Highly variable
• Depends mainly on:
• Severity of neurological involvement
• Tumor burden

Gardner syndrome is an autosomal dominant disorder characterized by colorectal polyposis and multiple extracolonic tumors.
It is considered a variant of Familial Adenomatous Polyposis (FAP).
• Caused by mutation in the APC gene on chromosome 5q21.
• This mutation leads to activation of the Wnt signaling pathway, resulting in uncontrolled cellular proliferation and tumor formation.
• Epidermoid cysts
• Common on face, scalp, and trunk
• Fibromas
• Lipomas
• Desmoid tumors (aggressive fibromatosis)
• May cause intestinal obstruction
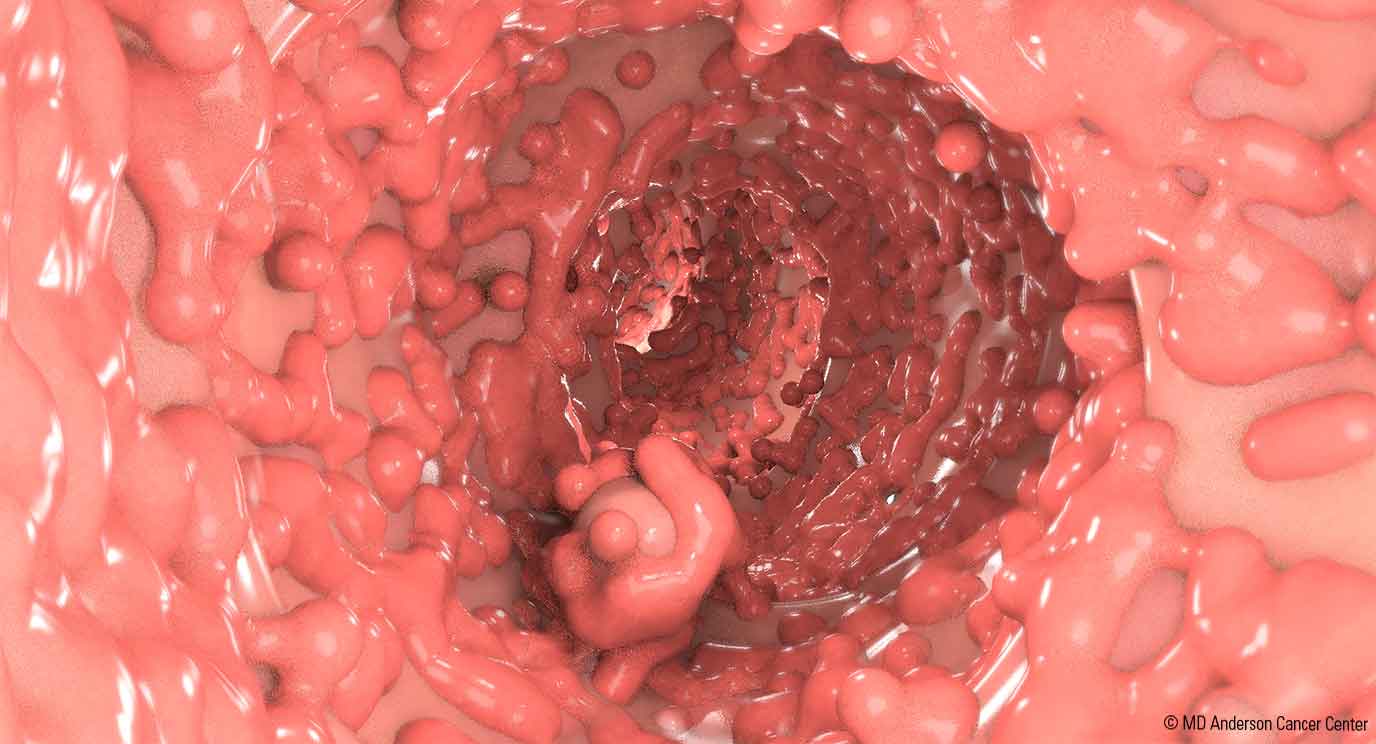
• Multiple adenomatous polyps of the colon
• If untreated, nearly 100% risk of colorectal carcinoma
• Upper gastrointestinal polyps
• Stomach
• Duodenum
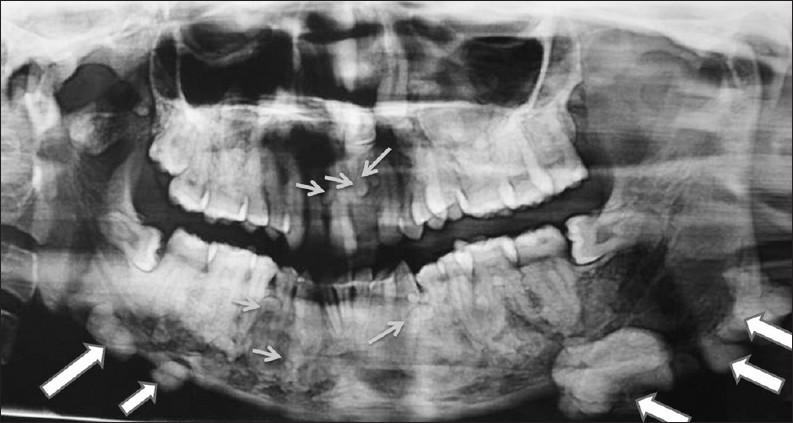
• Osteomas
• Commonly involve the skull and mandible
• Supernumerary teeth
• Papillary thyroid carcinoma
• Hepatoblastoma
• Genetic testing for APC mutation
• Colonoscopy
• Reveals numerous adenomatous polyps
• Panoramic dental X-ray
• Shows osteomas and supernumerary teeth
• Prophylactic colectomy
• Recommended due to near-certain risk of colorectal cancer
• NSAIDs
• Sulindac
• Celecoxib
• May reduce polyp burden
• Regular screening
• For extracolonic tumors
• Without surgical intervention, patients have very high mortality from colorectal carcinoma.
• Early diagnosis and prophylactic colectomy significantly improve survival.

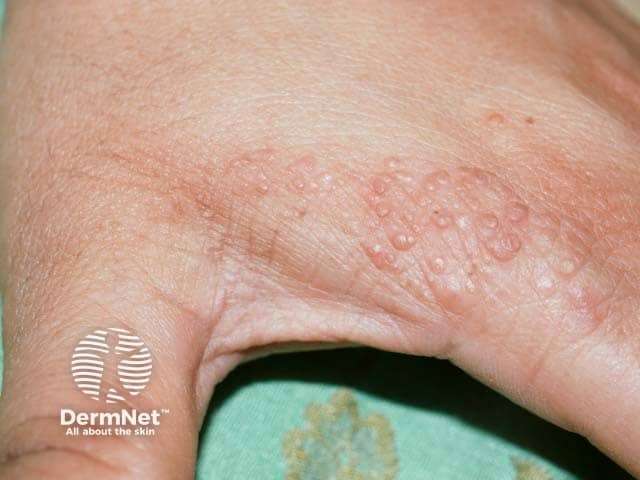

Cowden syndrome is an autosomal dominant genetic disorder characterized by multiple hamartomas involving several organ systems and a markedly increased risk of malignancies.
It belongs to the group of disorders known as PTEN Hamartoma Tumor Syndromes (PHTS).
• Caused by mutation in the PTEN gene located on chromosome 10q23.
• PTEN functions as a tumor suppressor gene that regulates cell proliferation.
• Mutation results in dysregulation of the PI3K/AKT/mTOR signaling pathway, leading to uncontrolled cellular growth and hamartoma formation.
• Facial trichilemmomas
• Benign tumors originating from hair follicles
• Acral keratoses
• Small wart-like lesions on hands and feet
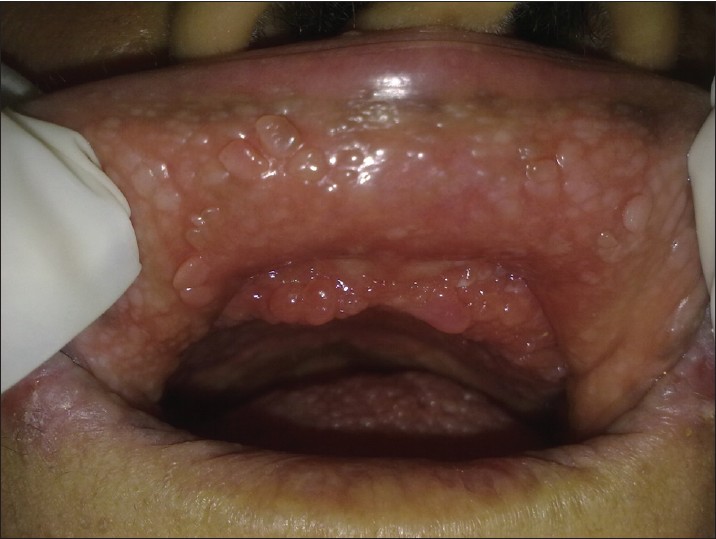
• Oral papillomas
• Produce a characteristic cobblestone appearance of the oral mucosa
• Breast cancer
• Approximately 85% lifetime risk
• Fibrocystic breast disease
• Endometrial carcinoma
• Approximately 30% lifetime risk
• Increased risk of follicular thyroid carcinoma
• Goiter
• Multiple thyroid nodules
• Hamartomatous gastrointestinal polyps
• Increased risk of colorectal cancer
Pathognomonic Features
• Facial trichilemmomas
• Oral papillomas
• Acral keratoses
Major Criteria
• Breast carcinoma
• Thyroid carcinoma
• Endometrial carcinoma
• Macrocephaly
• Lhermitte–Duclos disease
(dysplastic cerebellar gangliocytoma)
Minor Criteria
• Gastrointestinal hamartomas
• Fibromas
• Lipomas
Because of the high cancer risk, lifelong surveillance is essential.
• Annual breast MRI and mammography
• Transvaginal ultrasound
• Gynecological evaluation
• Regular thyroid ultrasound
• Periodic colonoscopy
• Monitoring for hamartomatous polyps
• Patients have a high lifetime risk of malignancies.
• Early detection and continuous screening significantly improve survival outcomes.
• Caused by TSC1 or TSC2 mutations, leading to mTOR pathway dysregulation.
• Characteristic skin findings:
• Facial angiofibromas
• Ash-leaf macules
• Shagreen patches
• Periungual fibromas
• Neurological manifestations:
• Seizures
• Cortical tubers
• Subependymal giant cell astrocytomas (SEGAs)
• Other systemic features:
• Renal angiomyolipomas
• Pulmonary lymphangioleiomyomatosis (LAM)
• Everolimus (mTOR inhibitor) is used for tumor control.
• Variant of Familial Adenomatous Polyposis (FAP) caused by APC gene mutation (5q21).
• Characterized by:
• Multiple colonic adenomatous polyps
• Nearly 100% risk of colorectal cancer
• Associated findings:
• Osteomas
• Epidermoid cysts
• Desmoid tumors
• Prophylactic colectomy is essential to prevent colorectal carcinoma.
• Caused by PTEN gene mutation (chromosome 10q23).
• Characterized by hamartomas with high malignancy risk.
Major cancer risks:
• Breast cancer (~85%)
• Thyroid carcinoma (follicular type)
• Endometrial carcinoma
Common cutaneous findings:
• Trichilemmomas
• Oral papillomas
• Gastrointestinal polyps
• Requires lifelong cancer surveillance and multidisciplinary management.
Biology has a recurring trick: when the brakes on cell growth fail, the body produces clusters of benign chaos—hamartomas. Syndromes like TSC, Gardner, and Cowden are different versions of the same cellular story: growth signals that should be carefully regulated begin whispering “grow” a little too loudly. Dermatology becomes the visible surface of those molecular whispers. Skin lesions often appear years before the internal cancers they warn about, making the skin a remarkably honest diagnostic messenger.
Get the full PDF version of this chapter.